Note
Click here to download the full example code
performing filtering using besca¶
This example demonstrates the entire process of filtering out cells/genes ob subpar quality before proceeding with analysis.
import besca as bc
import scanpy as sc
import matplotlib.pyplot as plt
import pytest
# pytest.skip('Test is only for here as example and should not be executed')
# load example dataset
adata = bc.datasets.pbmc3k_raw()
# set standard filtering parameters
min_genes = 600
min_cells = 2
min_UMI = 600
max_UMI = 6500
max_mito = 0.05
max_genes = 1900
visualization of thresholds¶
First the chosen thresholds are visualized to ensure that a suitable cutoff has been chosen.
# Visualize filtering thresholds
fig, ((ax1, ax2, ax3), (ax4, ax5, ax6)) = plt.subplots(ncols=3, nrows=2)
fig.set_figwidth(15)
fig.set_figheight(8)
fig.tight_layout(pad=4.5)
bc.pl.kp_genes(adata, min_genes=min_genes, ax=ax1)
bc.pl.kp_cells(adata, min_cells=min_cells, ax=ax2)
bc.pl.kp_counts(adata, min_counts=min_UMI, ax=ax3)
bc.pl.max_counts(adata, max_counts=max_UMI, ax=ax4)
bc.pl.max_mito(
adata, max_mito=max_mito, annotation_type="SYMBOL", species="human", ax=ax5
)
bc.pl.max_genes(adata, max_genes=max_genes)
![Expressed genes [count > 0], Cells that express a gene [count > 0], Counts per cell, max counts cutoff, mitochondrial gene content in dataset before filtering, Filtering by the maximum gene count](../../_images/sphx_glr_plot_example_filtering_001.png)
adding percent mitochondrial genes to dataframe for species human
application of filtering thresholds¶
Using the chosen thresholds the data is filtered. Before and after filtering results are depicted to compare.
# visualize data before filtering
sc.pl.violin(
adata, ["n_counts", "n_genes", "percent_mito"], multi_panel=True, jitter=0.4
)
print(
"The AnnData object currently contains:",
str(adata.shape[0]),
"cells and",
str(adata.shape[1]),
"genes",
)
print(adata)
# perform filtering
adata = bc.pp.filter(
adata,
max_counts=max_UMI,
max_genes=max_genes,
max_mito=max_mito,
min_genes=min_genes,
min_counts=min_UMI,
min_cells=min_cells,
)
# visualize data after filtering
sc.pl.violin(
adata, ["n_counts", "n_genes", "percent_mito"], multi_panel=True, jitter=0.4
)
print(
"The AnnData object now contains:",
str(adata.shape[0]),
"cells and",
str(adata.shape[1]),
"genes",
)
print(adata)
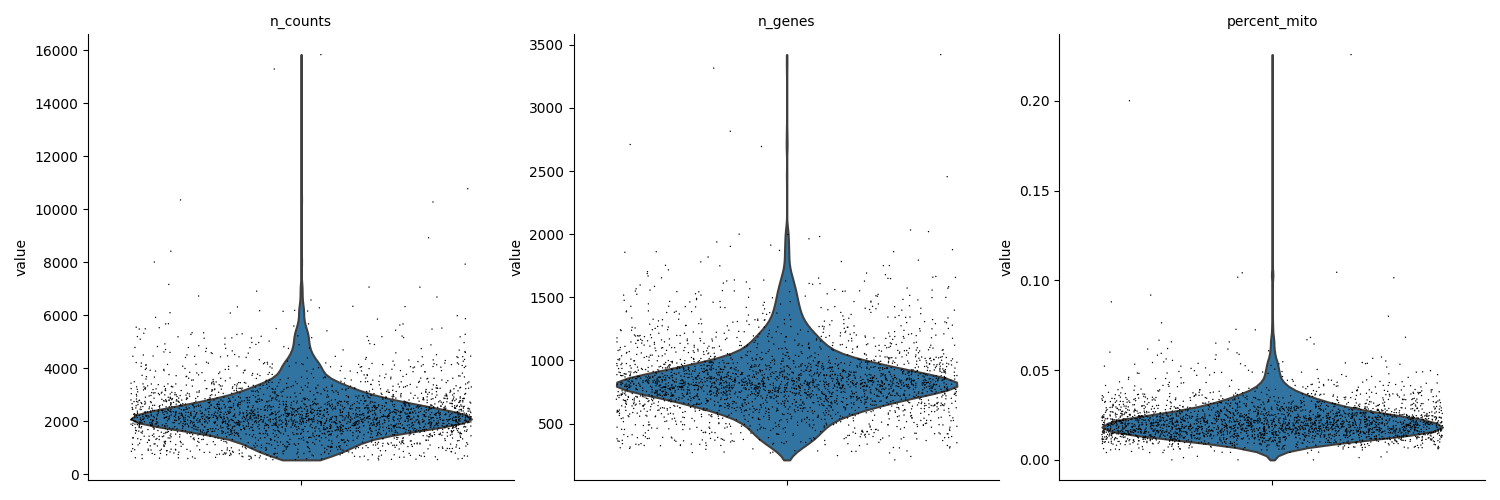
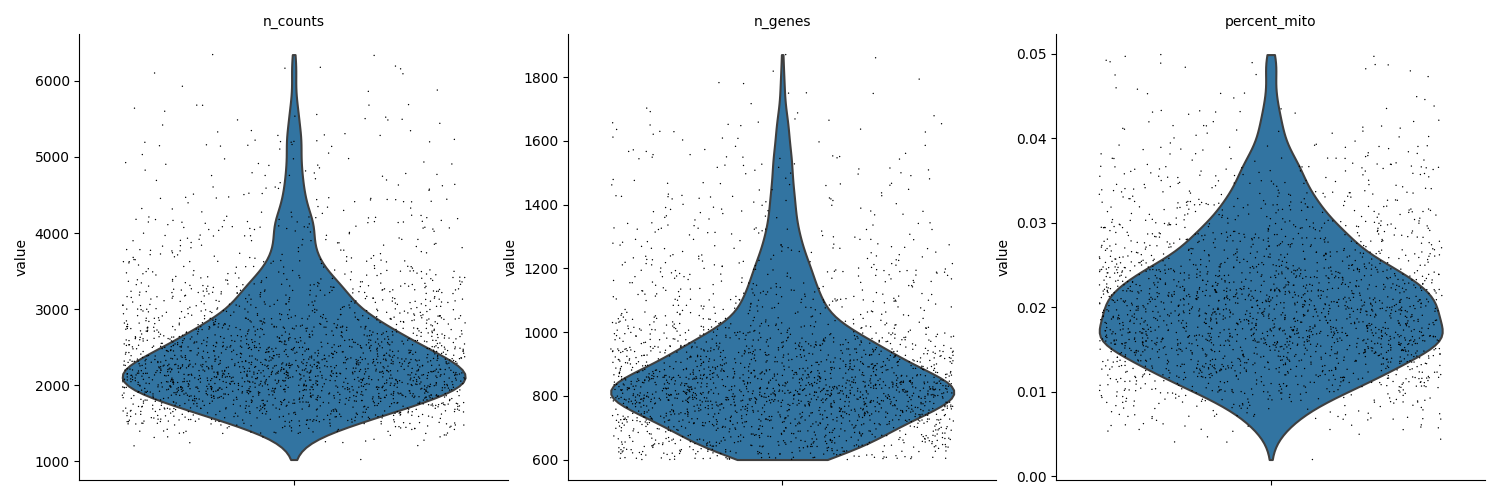
The AnnData object currently contains: 2700 cells and 32738 genes
AnnData object with n_obs × n_vars = 2700 × 32738
obs: 'CELL', 'n_counts', 'n_genes', 'percent_mito'
var: 'ENSEMBL', 'SYMBOL'
The AnnData object now contains: 2279 cells and 14702 genes
AnnData object with n_obs × n_vars = 2279 × 14702
obs: 'CELL', 'n_counts', 'n_genes', 'percent_mito'
var: 'ENSEMBL', 'SYMBOL', 'n_cells'
Total running time of the script: ( 0 minutes 0.964 seconds)